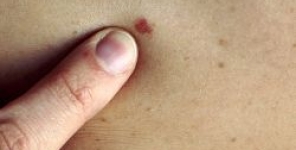
Al vaglio dell'FDA le iniezioni di T-VEC per la terapia del melanoma
Un trattamento sperimentale per il melanoma, da iniettare direttamente nella lesione, è al vaglio degli esperti dell'FDA per ottenere l'approvazione all'impiego.
Il prodotto in questione, il talimogene laherparepavec (T-VEC, della Amgen Inc), viene descritto dall'azienda produttrice come il primo tipo di immunoterapia oncolitica ed è destinato all'impiego in pazienti con lesioni da melanoma iniettabili.
Contiene una versione geneticamente modificata del virus herpes simplex che è stata manipolata per replicarsi selettivamente all'interno del tumore e provocare la distruzione delle cellule tumorali. Il prodotto contiene anche un gene per il fattore di crescita dei granulociti e dei macrofagi (GM-CSF), che viene espresso localmente nel tumore. La rottura delle cellule tumorali causa il rilascio di antigeni tumorali derivati che, in sinergia con il GM-CSF rilasciato dal farmaco, stimolano una risposta immunitaria sistemica.
La raccomandazione per l'approvazione da parte dell'FDA, passata con una votazione di 22 voti a favore contro 1 voto contrario, è il risultato della riunione di due comitati consultivi, l'Oncologic Drugs Advisory Committeeed il Cellular, Tissue and Gene Therapies Advisory Committee.
Il risultato della votazione è stato inusuale, considerato che il documento informativo rilasciato dall'FDA prima della riunione conteneva una lunga lista di interrogativi ancora aperti e poneva la questione dell'accettabilità del profilo del farmaco in termini di rischi-benefici, in un campo terapeutico in cui sono stati recentemente presentate molte nuove terapie.
Il campo della cura del melanoma ha infatti visto succedersi molti nuovi prodotti nel corso degli ultimi anni, tra cui immunoterapie basate su inibitori di check-point e almeno un inibitore di mutazione genetica (mirato a BRAF e MEK); tutti questi nuovi farmaci hanno mostrato di poter ottenere un miglioramento statisticamente significativo della sopravvivenza globale rispetto alla terapia standard.
Nei dati derivati dagli studi clinici presentati nel corso della riunione sul talimogene laherparepvec, i dati di sopravvivenza globale (un endpoint secondario, nel contesto della valutazione) hanno mancato di poco il traguardo della significatività statistica, e il vantaggio che ha mostrato rilievo statistico era parte di un endpoint composito che ha reso la valutazione "poco chiara", nei termini dei revisori dell'FDA che hanno commentato il documento informativo.
L'FDA prevede di arrivare ad una decisione definitiva sull'approvazione del talimogene laherparepavec prima della fine di ottobre 2015.
Trial clinici
I dati clinici presentati per l'approvazione provengono principalmente da uno studio di fase 3 noto come Optim (o studio 005/05). Lo studio ha coinvolto 436 pazienti con melanoma maligno non asportabile chirurgicamente, assegnati randomicamente (nella proporzione 2:1) al gruppo talimogene laherparepavec o al gruppo controllo (GM-CSF). Il trattamento è stato iniettato direttamente in tutte le lesioni cutanee ragionevolmente iniettabili.
La dose iniziale di talimogene laherparepvec era pari a 4 mL totali, ad una concentrazione di 106 PFU / mL. Dosi successive sono state somministrate 3 settimane dopo la prima dose e consistevano di talimogene laherparepvec (fino a 4 mL totali, concentrazione di 108 PFU / mL) ogni 2 settimane.
Il controllo (GM-CSF) è stato somministrato alla dose di 125 ug / m2 / die per via sottocutanea, con una tabella clinica di 4 settimane, suddivisa in 14 giorni a dose giornaliera e 14 giorni di sospensione.
L'endpoint primario era il tasso di risposta durevole, definito come la percentuale di pazienti che hanno manifestato una risposta completa o parziale (CR o PR) iniziata in qualsiasi momento entro 12 mesi dall'inizio della terapia e mantenuta per almeno 6 mesi. Questi risultati mostrano un vantaggio significativo del prodotto in esame, con un tasso di risposta durevole del 16,3% per il braccio talimogene laherparepvec rispetto al 2,1% del controllo (P < .0001).
La sopravvivenza generale è stato un endpoint secondario. Ci sono state 189/295 (64%) morti confermate nel gruppo talimogene laherparepvec e 101/141 (72%) nel gruppo controllo (P = .051). Le stime in termini di sopravvivenza totale media (OS) erano di 23,3 mesi per il braccio talimogene laherparepvec e di 18,9 mesi per il braccio controllo (rapporto di rischio 0,79).
Gli effetti collaterali più frequenti collegati al trattamento con talimogene laherparepvec sono stati, secondo i documenti informativi FDA: affaticamento, brividi, febbre, nausea, sintomi influenzali e dolore al sito di iniezione. Sintomi influenzali sono stati riportati dal 90% dei pazienti.
Un importante effetto collaterale è stata la cellulite al sito di iniezione, causata dal metodo di somministrazione, l'iniezione intratumorale, del talimogene laherparepvec. Nel braccio laherparepvec talimogene, 18 pazienti (6,2%) hanno sviluppato cellulite; fra questi, 7 eventi (2,4%) sono stati classificati come gravi, al punto da richiedere il ricovero ospedaliero. Un paziente ha sviluppato una ferita che è diventata resistente alla terapia, tanto che si è reso necessario sottoporre il paziente ad amputazione della gamba al di sotto del ginocchio; i revisori dell'FDA hanno tuttavia sottolineato che il rapporto fra questa complicanza e la somministrazione di talimogene laherparepvec non è del tutto chiaro.