Algoritmo di gestione della porfiria eritropoietica congenita, derivato da uno studio di 29 casi
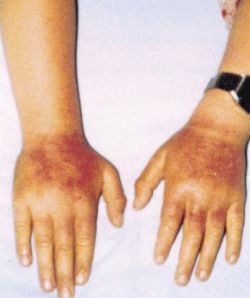 La porfiria eritropoietica congenita (CEP) è una porfiria autosomica recessiva foto-mutilante, di solito con insorgenza durante l'infanzia, nella quale le complicanze ematologiche determinano la prognosi. A causa della sua estrema rarità e della sua eterogeneità clinica, le decisioni di gestione della CEP sono spesso difficili.
La porfiria eritropoietica congenita (CEP) è una porfiria autosomica recessiva foto-mutilante, di solito con insorgenza durante l'infanzia, nella quale le complicanze ematologiche determinano la prognosi. A causa della sua estrema rarità e della sua eterogeneità clinica, le decisioni di gestione della CEP sono spesso difficili.
Obiettivi: Sviluppare un algoritmo di gestione per i pazienti affetti da CEP, basato sui dati provenienti da anamnesi attentamente caratterizzate.
Metodi: Una singola persona ha raccolto i dati relativi ai trattamenti, e ai loro risultati, di 29 pazienti con CEP da Regno Unito, Francia, Germania e Svizzera.
Risultati: Sei bambini sono stati sottoposti a trapianto di midollo osseo (BMT); cinque non hanno presentato sintomi fino a 11.5 anni dopo il trapianto. I trattamenti come il carbone per via orale, la splenectomia e l'iper-trasfusione cronica non hanno dato alcun beneficio o addirittura hanno portato a complicazioni e ad un impatto negativo sulla qualità della vita legata alla salute (HRQoL). La mancanza di una coerente correlazione tra genotipo e fenotipo sta a significare che questa non può essere usata per prevedere la prognosi della malattia. I principali fattori per una prognosi sfavorevole sono stati la giovane età di insorgenza della malattia e la gravità delle manifestazioni ematologiche.
Conclusioni: È stato proposto un algoritmo di gestione in cui ogni paziente, a prescindere dalla gravità della malattia al momento della presentazione, dovrebbe ricevere una valutazione clinica globale e multidisciplinare e dovrebbe essere riesaminato ad intervalli in base alla sua prognosi prevista e al tasso di insorgenza delle complicazioni. Dovrebbe essere considerato il BMT per quei pazienti con trombocitopenia e/o anemia emolitica sintomatica progressiva. I genotipi dell'uroporfirinogeno III sintasi associati a prognosi sfavorevole dovrebbero inoltre giustificare la considerazione per un BMT. Una rigorosa foto-protezione della pelle e degli occhi dalla luce visibile è comunque essenziale in tutti i pazienti.
Storia della pubblicazione:
Titolo: A management algorithm for congenital erythropoietic porphyria derived from a study of 29 cases.
Rivista: British Journal of Dermatology. doi: 10.1111/j.1365-2133.2012.11154.x
Autori: RP. Katugampola, AV. Anstey, AY. Finlay, S. Whatley, J. Woolf, N. Mason, JC. Deybach, H. Puy, C. Ged, H. deVerneuil, S. Hanneken, E. Minder, X. Schneider-Yin, MN. Badminton
Affiliazioni: Department of Dermatology and Wound Healing, Cardiff University, UK
Department of Medical Biochemistry and Immunology, University Hospital of Wales, Cardiff, UK
INSERM U773, Centre de Recherche Biomédicale Bichat-Beaujon, Université Paris Diderot, site Bichat, 75018 Paris, France and AP-HP, Centre Français des Porphyries, Hôpital Louis Mourier, Colombre, France
INSERM U1035, Bordeaux-Segalen University, Bordeaux, France
Department of Dermatology, University Hospital of Duesseldorf, Germany
Institute of Laboratory Medicine, Stadtspital Triemli, Zürich, Switzerland.
Abstract:
Background: Congenital Erythropoietic Porphyria (CEP) is an autosomal recessive photo-mutilating porphyria with onset usually in childhood, where haematological complications determine prognosis. Due to its extreme rarity and clinical heterogeneity management decisions in CEP are often difficult.
Objectives: To develop a management algorithm for patients with CEP based on data from carefully characterised historical cases.
Methods: A single investigator collated data related to treatments and their outcomes in 29 CEP patients from the UK, France, Germany and Switzerland.
emoliaResults: Six children were treated with bone marrow transplantation (BMT); five have remained symptomatically cured up to 11.5 years post-transplantation. Treatments such as oral charcoal, splenectomy and chronic hyper-transfusion were either of no benefit or associated with complications and negative impact on health-related quality of life (HRQoL). Lack of consistent genotype-phenotype correlation meant that this could not be used to predict disease prognosis. The main poor prognostic factors were early age of disease onset and severity of haematological manifestations.
Conclusions: A management algorithm is proposed where every patient, irrespective of disease severity at presentation, should receive a comprehensive, multi-disciplinary clinical assessment and should then be reviewed at intervals based on their predicted prognosis, and the rate of onset of complications. A BMT should be considered in those with progressive, symptomatic haemolytic anaemia and/or thrombocytopenia. Uroporphyrinogen III synthase genotypes associated with poor prognosis would additionally justify consideration for a BMT. Rigorous photo-protection of skin and eyes from visible light is essential in all patients.





